

近日,百蓁生物创始人李明院士带领加拿大滑铁卢大学、百蓁生物、清华大学、中原AI院联合团队攻关,在国际期刊《Nature Biotechnology》发表题为“Zero-Shot De Novo Peptide Sequencing with Open Posttranslational Modification Discovery”的研究成果,该项研究成果首次实现了无需预定义修饰列表、无需重新训练的零样本开放翻译后修饰(PTM)从头测序,解决了深度学习蛋白测序长期被 “封闭世界假设” 限制的核心痛点,将大大推进“暗蛋白质组”(dark proteome)的系统性探索研究工作。百蓁生物参与合作完成了该项研究的算法湿实验验证等相关工作。 ▶ 发表时间:2026年5月19日 ▶ 期刊:《Nature Biotechnology》(IF=41.7) ▶ 文章标题:Zero-Shot De Novo Peptide Sequencing with Open Posttranslational Modification Discovery 在蛋白质组学中,从串联质谱数据中鉴定肽段序列是一项核心任务。传统方法——数据库搜索(Database Search)依赖蛋白质数据库,无法识别未知剪接变体,如未注释的开放阅读框(ORF),稀有或未报道的翻译后修饰(PTMs)等。深度学习显著提升了从头测序的准确性,但仍受限于封闭的氨基酸与修饰集合,如现有深度学习de novo模型(如GraphNovo、CasaNovo、InstaNovo、PrimeNovo):虽然显著提升了标准肽段的测序精度,但均受限于"封闭世界假设"——它们只能在训练时预定义的氨基酸/修饰词汇表内工作,无法泛化到未知修饰。对于训练时未见过的新型或稀有PTM,这些模型完全失效。 开放PTM发现是一个理论上"无边界"的问题:可能的化学修饰空间无限;修饰在质谱图中的信号往往微弱,且与已知残基或噪声重叠;不同于标准测序(在20种标准氨基酸的封闭离散词汇表上操作),开放发现需要在无候选列表、无预定义规则的情况下,对未知化学组成进行推理。 传统开源搜库(如 MSFragger Open Search)灵敏度极低,会漏检大量真实修饰。本文要解决的问题:如何让 AI 模型在零样本条件下,自动发现从未见过、未定义的翻译后修饰,并完成高精度从头测序。 本研究提出 RNoVA(Rotary positional embedding-enhanced de Novo sequencing Algorithm),一个基于Transformer的模块化框架,能够在零样本条件下同时完成高精度肽段测序和开放PTM发现,无需重新训练或预设修饰列表。这篇论文首次实现了无需预定义修饰列表、无需重新训练的零样本开放翻译后修饰(PTM)从头测序,解决了深度学习蛋白测序长期被 “封闭世界假设” 限制的核心痛点,是质谱蛋白质组学与 AI 结合的里程碑工作。 RNoVA 提出了一个模块化、基于Transformer的框架,采取了将修饰检测(PathSearcher)与序列推断(SeqFiller)分离的解耦策略;RNovA由两个独立训练的Transformer模块组成(均为6层、768维隐藏层、12个注意力头),通过RoPE和Flash Attention增强。 图1:RNoVA架构与工作流程 PathSearcher:将MS/MS串联质谱建模为图,节点为碎片离子峰,在质谱图中搜索最优碎片离子路径,输出质量标签序列(mass tags),其中可能包含未知修饰的质量偏移。 输出意义:路径中的相邻节点质量差即为质量标签(mass tags)。若质量差匹配标准氨基酸质量,则直接翻译为残基;若不匹配,则保留为未解析的质量标签(可能代表多个氨基酸的组合质量,或携带未知PTM的单个残基)。因此,PathSearcher能在无任何先验假设的情况下检测新型修饰。 SeqFiller:根据已生成的肽段前缀(prefix)、用户定义的候选氨基酸列表(可包含标准氨基酸+候选修饰残基,如K[+42.011])以及前缀质量嵌入(prefix mass embedding),将质量标签序列转化为完整的氨基酸序列,支持用户自定义候选氨基酸(包括未知修饰),无需重新训练。 零样本特性:虽然SeqFiller依赖用户提供的候选列表,但它是完全零样本的——即使候选列表包含训练时未见过的PTM残基,也无需重新训练或微调即可直接部署。 零样本泛化:利用旋转位置编码(RoPE)捕捉碎片离子间的质量差异,而非绝对质量值,从而泛化到未见过的肽段化学修饰。 使用序列级奖励信号而非token级监督,增强模型对缺失碎片、噪声和未知修饰的鲁棒性。 训练过程中使用Q-learning和价值函数,鼓励探索全局一致的序列路径。强化学习式训练:将测序视为序列决策问题,使用序列级反馈而非残基层监督,容忍因碎片缺失或未知修饰导致的模糊性。 全自动端到端流程:从质谱图→质量标签聚类→PTM推断→完整序列重建,无需预定义修饰列表或重新训练。自动从PathSearcher输出的质量标签中聚类、筛选富集的修饰候选,传递给SeqFiller进行完整序列重建。无需人工干预,不依赖数据库。通过自动化的质量标签聚类与富集分析,RNoVA可直接从数据中发现统计学显著的PTM候选。 RNoVA为蛋白质组中“暗物质”的探索提供了强大工具,尤其在免疫肽组学、临床标志物发现和非模式生物研究中具有广泛应用前景。

RNoVA 的核心创新
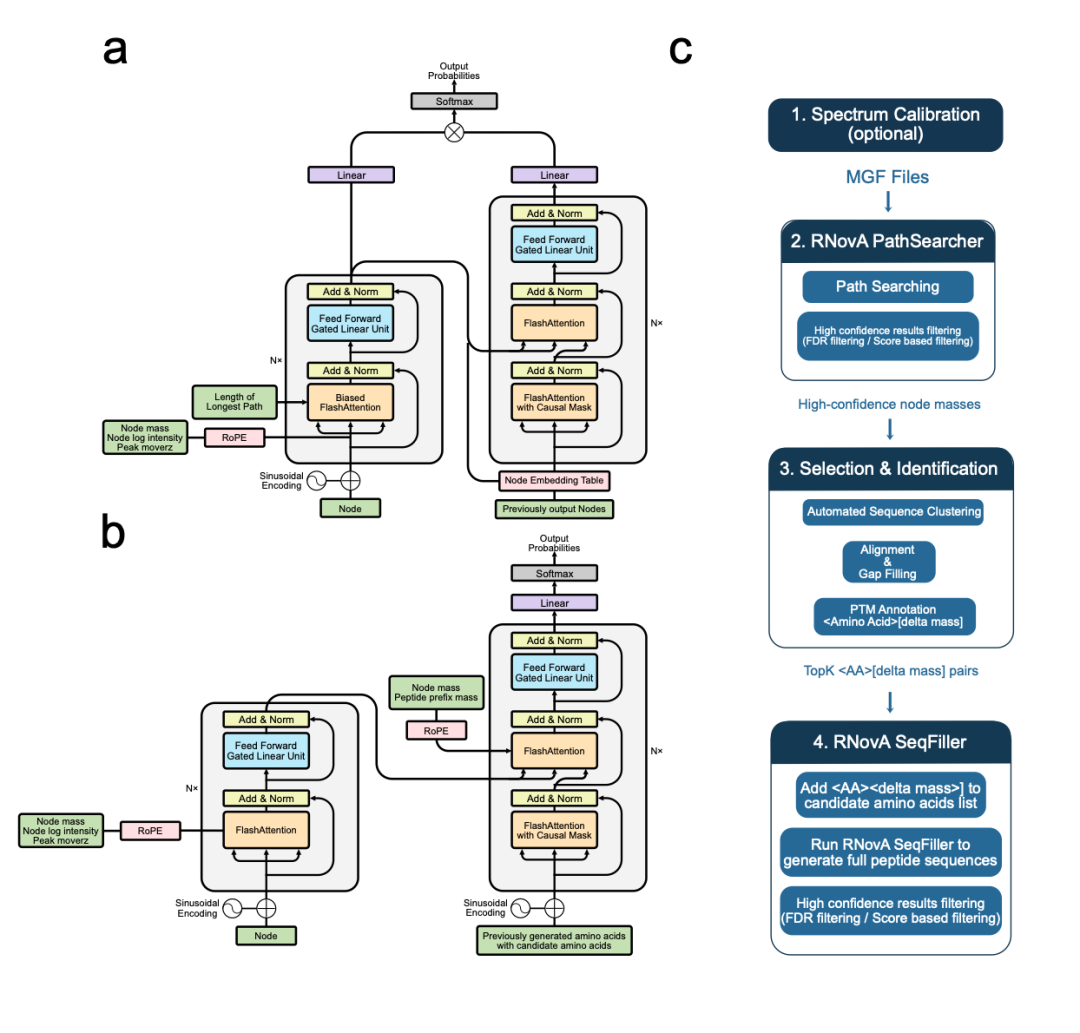
核心创新内容
1. 双模块设计
2. 旋转位置编码(RoPE)
3. 强化学习风格的训练框架
4. 自动化的开放PTM发现流程
实验结果与验证
1. 标准数据集上的SOTA性能
在三个模式生物(拟南芥、秀丽隐杆线虫、大肠杆菌)上,RNoVA在肽段召回率(RNovA 79.17%–82.11%)、氨基酸召回率/精确率上,均超 GraphNovo、CasaNovoV2、InstaNovo、PrimeNovo达到最优。在节点级别的碎片离子上,与GraphNovo持平或略优,尽管RNoVA搜索空间更大(允许任意质量差)。RNovA在开放搜索空间下的节点级性能与封闭模型相当,证明了RoPE增强架构的鲁棒性。
图2:标准数据集上的性能测试比对 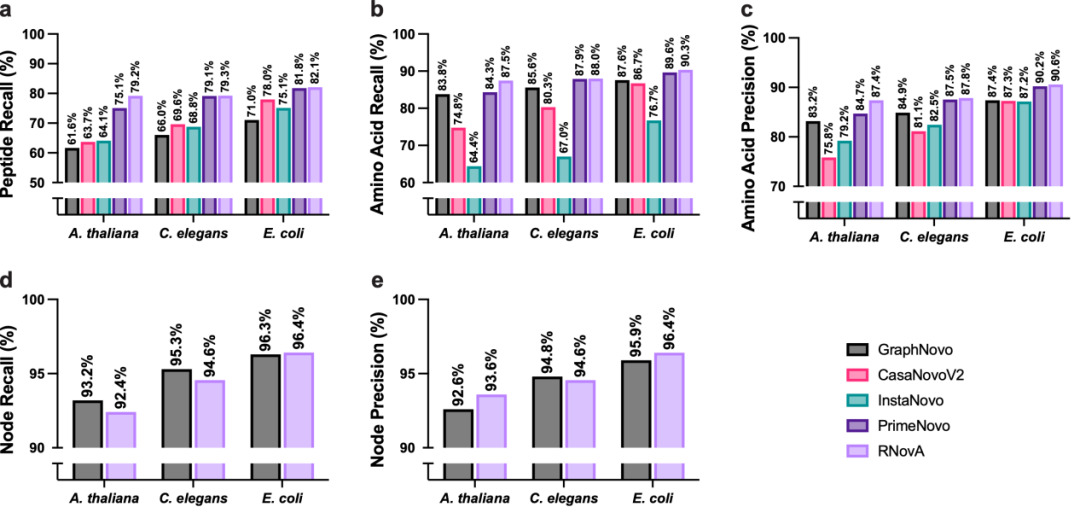
2. RNoVA在零样本PTM鉴定中的系统性优势
在ProteomeTools数据集(PXD009449)上,RNoVA展现出对传统de novo工具PEAKS的全面超越。该数据集涵盖磷酸化、乙酰化、巴豆酰化、琥珀酰化、瓜氨酸化等21种翻译后修饰(PTM),其中仅乙酰化(K_Acetyl)和酪氨酸磷酸化(Y_Phospho)见于RNoVA训练集,其余19种均为零样本识别。
氨基酸水平性能:RNoVA在召回率与精确率上均显著优于PEAKS。典型地,Y_Phospho精确率达95.44%(PEAKS 86.87%),巴豆酰化(K_Crotonyl)精确率达95.67%(PEAKS 83.92%)——即使后者为模型训练阶段从未见过的修饰类型。
修饰位点定位精度:RNoVA的PathSearcher模块可独立实现高置信度位点定位,PTM位点定位准确率平均提升>10个百分点。例如Y_Phospho位点定位准确率达88.46%,戊二酰化(K_Glutaryl)达93.63%,且修饰残基上的性能衰减幅度远小于PEAKS。
修饰残基性能鲁棒性:PEAKS在修饰位点的召回率较未修饰位点下降超10个百分点;而RNoVA的性能衰减显著更小,在Y_Phospho和K_Crotonyl等修饰上,其对修饰残基的召回率甚至超过了PEAKS对未修饰残基的表现——表明模型对PTM的识别能力已达到超越传统工具对普通氨基酸的识别水平。
图3:对多种PTM的零样本泛化分析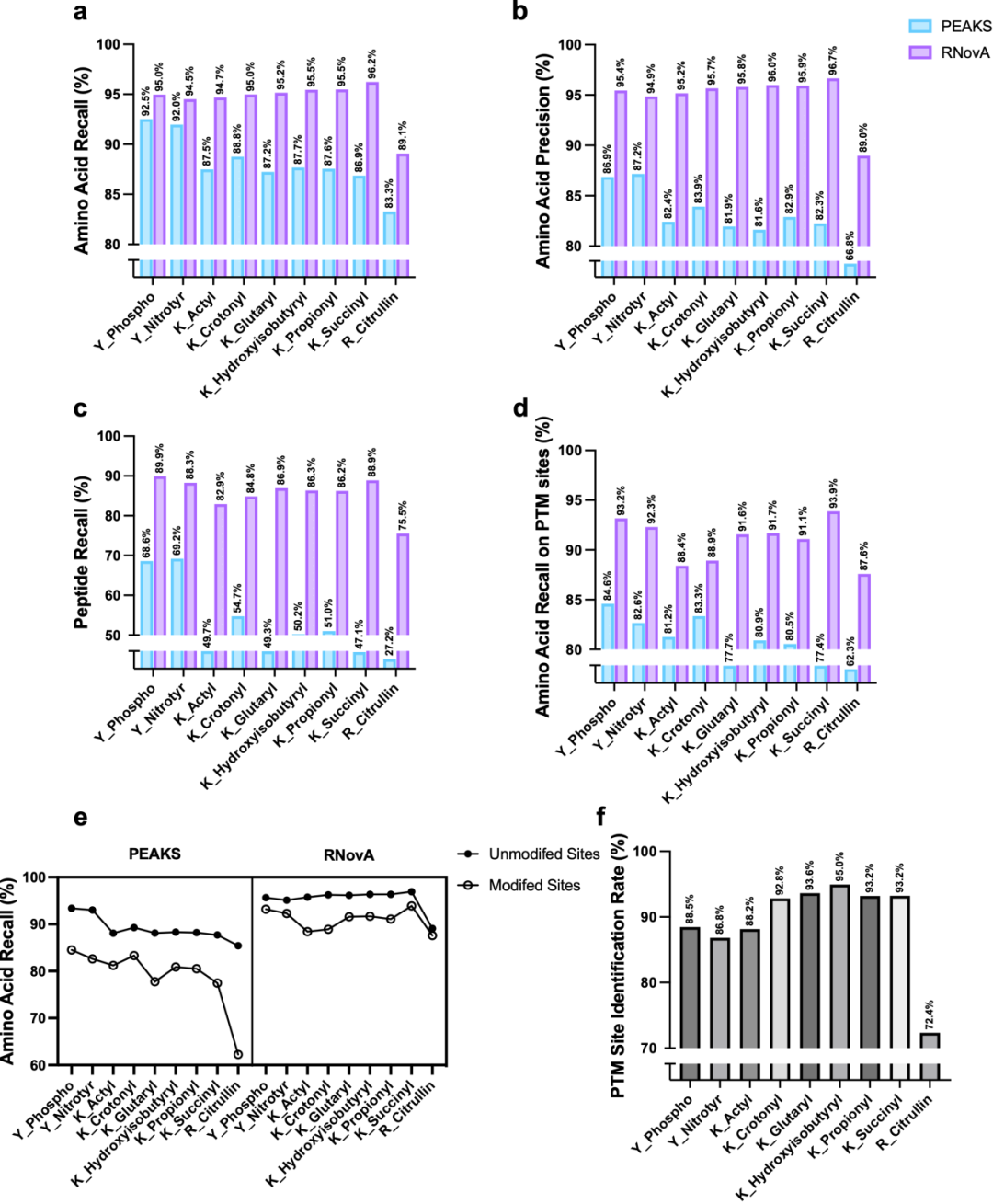
3. 无数据库FDR控制策略
针对de novo鉴定缺乏目标-诱饵数据库对照的问题,我们设计了基于谱图扰动的FDR估计框架:
诱饵谱图生成:对每个实验谱图,随机移除40%信号峰并替换为同分布噪声峰,生成保留原始统计特性的诱饵谱图。
目标-诱饵竞争:每个扫描循环中,仅保留目标谱图与诱饵谱图得分更高的PSM(Peptide-Spectrum Match)进入下游分析。
分层FDR估计:分别对PathSearcher(节点级,即修饰位点/氨基酸残基水平)和SeqFiller(氨基酸级,即序列组装水平)独立估计1% FDR阈值。
正交验证(¹⁵N同位素基准):采用¹⁵N标记酵母蛋白质组进行独立验证。经同位素修正后,PathSearcher实际FDR为1.7% ± 1.6%,SeqFiller为0.6% ± 0.4%,均与1%目标阈值高度一致,证明该无数据库FDR策略的可靠性。
图4:1515N同位素FDR验证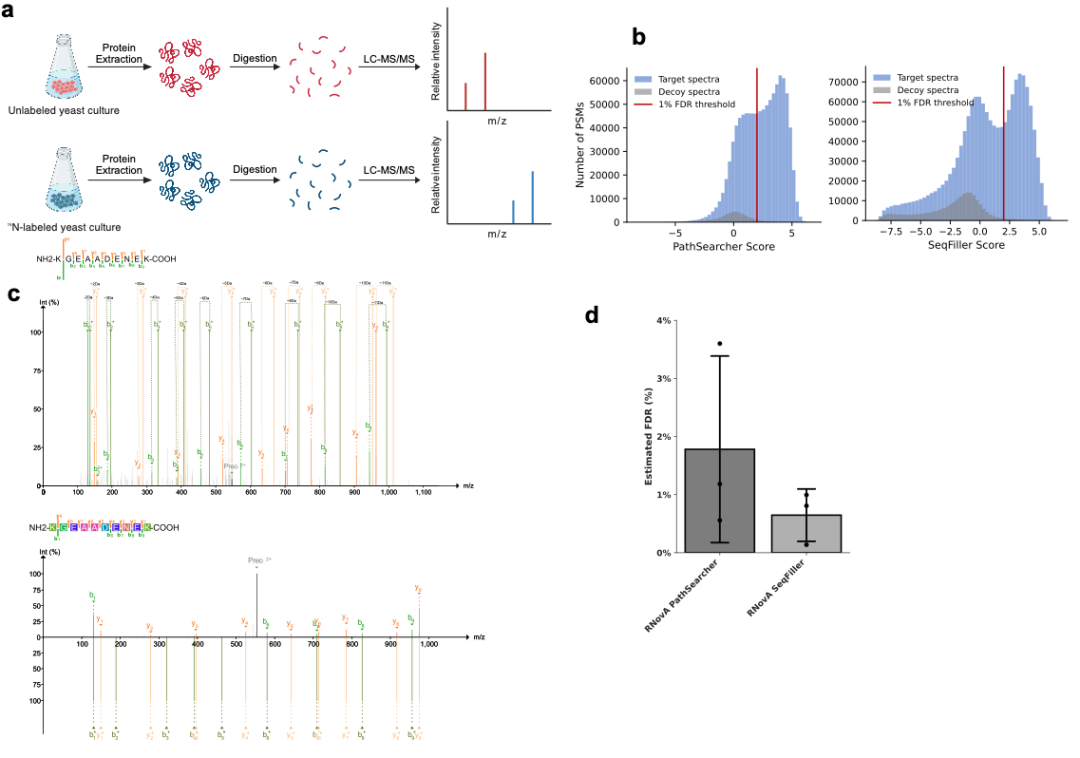
4. 合成非天然PTM的开放发现实验——人工合成修饰的极限验证
为评估RNoVA对未知修饰的发现能力,我们设计了4种自然界不存在的人工修饰(TestMod1–4),分别连接于赖氨酸(K[TestMod:1]、K[TestMod:2])、丝氨酸(S[TestMod:3])和丙氨酸(A[TestMod:4]),合成24条修饰肽段掺入293T细胞裂解液中。
PathSearcher:在1% FDR下成功鉴定全部4种合成PTM,获得48–498个高置信度PSM,实现对完全未知修饰类型的零样本发现。
对比方法:MSFragger开放搜索仅检出11个K[TestMod:2]的PSM,其余3种修饰完全失败——表明传统开放搜索策略对训练集外修饰的识别存在根本性局限。
SeqFiller序列重建:在复杂细胞背景下仍能完整重建修饰肽段序列,精确率>99%,召回率稳健。
浓度梯度鲁棒性:在400 fmol、40 fmol、4 fmol三级稀释系列中,精确率始终>99.9%。该结果表明,当修饰信号可被质谱采集到时,算法本身不构成灵敏度瓶颈。
图5:合成修饰肽解析能力验证
5. 类风湿关节炎中的新型修饰发现
本研究开展了对类风湿关节炎患者滑膜组织蛋白质组学数据的de novo分析,PathSearcher鉴定出Top 5质量偏移,其中色氨酸→犬尿氨酸(Kynurenine)氧化最为显著。从RA患者滑膜组织中直接鉴定到含犬尿氨酸的肽段提示色氨酸代谢通路在RA病理中的潜在作用。
为了验证该修饰的可靠性,研究团队合成了两条候选肽段进行LC-MS/MS分析,结果显示与内源性信号的保留时间(RT)高度一致,碎片谱图匹配良好,证实该修饰的真实存在。BLAST比对也显示,一条肽段完全匹配已知蛋白;另一条存在新型修饰的同时还存在单残基替换突变,提示可能的等位基因变异或蛋白异构体。
图6:RA样本中犬尿氨酸修饰的发现与验证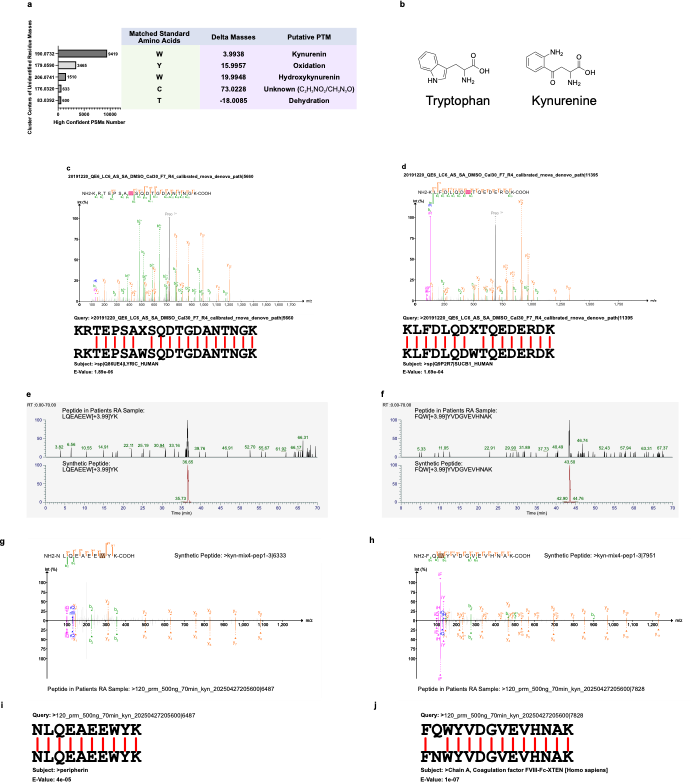
6. 完全无参考蛋白组条件下的新型PTM发现
对菌株A1232E(Nesterenkonia属,无参考蛋白质组数据库)进行de novo分析,PathSearcher发现富集质量偏移172.08 Da,推断为谷氨酸残基(129.04 Da)携带+43.04 Da未知修饰。
序列重建:SeqFiller将E[+43.04]纳入候选修饰集后,成功重建完整肽段序列。
化学推断:结合质量偏移与生物化学背景,鉴定该修饰为Ethanolamine, Eth。
合成验证:合成肽段LDDKLNNKAEEE[Eth]GATK,其保留时间(36.96 min vs 内源性37.78 min)和碎片谱图与内源性信号高度吻合,证实该修饰的真实存在。
该案例证明,RNoVA在完全缺乏参考数据库的条件下,仍可实现新型PTM的从头发现、化学推断与实验验证,突破了传统开放搜索对数据库依赖的根本限制。
图7:无参考蛋白组细菌中的新修饰发现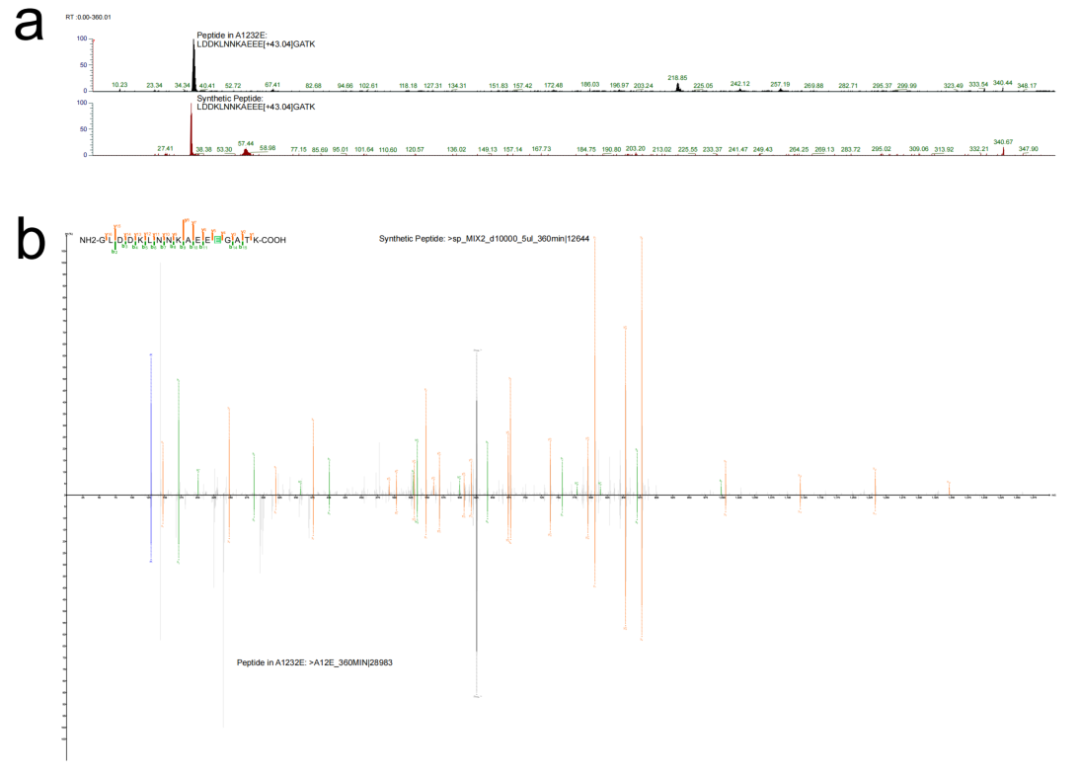
核心意义
打破封闭世界假设:首次实现无需重训练、无需候选列表的开放PTM发现;
模块化设计:PathSearcher负责无偏发现,SeqFiller负责精确重建,两者可独立使用或协同;
实用化FDR控制:NovoBoard框架为de novo预测提供了无数据库的置信度评估;
跨物种通用性:在无参考基因组的非模式生物中同样有效。
一句话结论
RNovA 用 Transformer + RoPE + 类强化学习,首次实现了零样本、无预设、无数据库的开放 PTM 从头测序,在标准测序、已知 PTM、全新人工修饰、临床样本、无参考基因组场景全面验证有效,是蛋白质组学 AI 从头测序的新一代基础框架。









