

关键词:
主动脉瘤和夹层 (AAD) 是一种致命的心血管急症,其发病迅速且致死率极高。疾病发生时,内膜撕裂导致血液进入中膜,破坏了血流动力学和血管壁的完整性。尽管影像学和外科学取得了显著进展,但目前临床上仍缺乏能够有效延缓或逆转这种退行性血管重构的内科药物。因此,阐明驱动 AAD 发病的核心分子机制,对于改善预后和创新治疗策略至关重要。
2026 年 6 月 4 日,复旦大学附属中山医院葛均波院士/任骏教授/张英梅教授团队在国际心血管顶级期刊 Circulation Research 上发表了题为「ATAD3A Limits Aortic Dissection via Mito-Lysosome Contacts and Lipoylation」的研究论文。该研究首次揭示了线粒体内膜蛋白 ATAD3A 在保护血管平滑肌细胞 (VSMC) 免受铜死亡 (cuproptosis) 中的关键作用,为 AAD 的治疗提供了全新的代谢干预靶点。
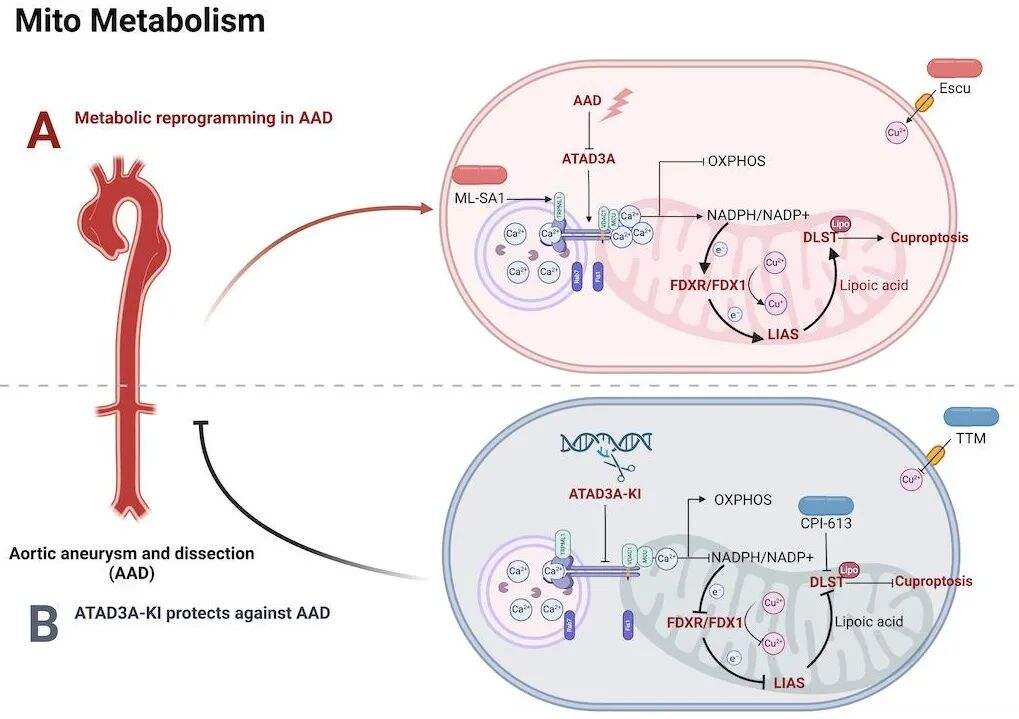
研究团队首先通过多组学交叉筛选,将目光锁定在线粒体 AAA ATP 酶-ATAD3A 上。进一步的组织学及表达谱分析发现,在人类胸主动脉夹层 (TAD) 样本以及 β-氨基丙腈 (BAPN) 诱导的 AAD 小鼠模型中,血管平滑肌中 ATAD3A 的表达水平均呈现出显著升高的趋势。
为明确 ATAD3A 在 AAD 病理过程中的具体影响,研究团队构建了全身性 ATAD3A 过表达敲入 (ATAD3A-KI) 小鼠模型。体内动物实验表明,ATAD3A 的过表达能够显著减轻 BAPN 和 AngII 诱导的主动脉扩张,大幅降低夹层的发生率,并切实改善了小鼠的整体生存率。相反,在 VSMC 特异性敲低 ATAD3A 的小鼠模型中,血管壁弹性纤维的断裂和病理损伤则显著加剧,从正反两面证实了内源性 ATAD3A 在限制 AAD 进展中发挥着不可或缺的保护作用。
在探究其深层分子机制时,团队通过转录组与蛋白组的联合分析发现,细胞器间的相互作用在维持血管壁代谢稳态中扮演着重要角色。在 AAD 的病理状态下,作为一种富集于细胞器接触位点的关键蛋白,ATAD3A 能够有效重塑线粒体-溶酶体接触 (MLC)。一方面,ATAD3A 过表达减少了异常的线粒体-溶酶体接触,进而通过阻断 TRPML1-VDAC1-MCU 信号轴,强效限制了线粒体钙离子的内流。这一机制不仅稳定了线粒体膜电位,还大幅降低了线粒体活性氧 (ROS) 的爆发和 NADPH 的过度消耗,维持了细胞的氧化还原稳态。
另一方面,伴随着钙信号的稳定,ATAD3A 进一步抑制了下游 FDXR/FDX1/LIAS 代谢通路的异常活跃状态。该作用直接削弱了三羧酸循环核心酶-二氢硫辛酰胺琥珀酰转移酶 (DLST) 的硫辛酰化修饰程度。由于高硫辛酰化的 DLST 极易与细胞内过量的铜离子结合并触发铜死亡级联反应,ATAD3A 正是通过降低 DLST 的硫辛酰化水平,成功阻断了 VSMC 的死亡进程,从而保护了血管平滑肌细胞的存活与收缩表型。此外,体内外拯救实验证实,靶向线粒体的代谢抑制剂 Devimistat (CPI-613) 能够有效抑制 DLST 的硫辛酰化及随后的铜死亡,显著减轻主动脉夹层的严重程度并降低破裂死亡率。这为未来预防和靶向治疗主动脉夹层提供了一种极具潜力的高效临床干预策略。
综上,本研究揭示了 ATAD3A 是主动脉疾病中重要的对抗铜死亡调控节点,其通过调控线粒体-溶酶体接触限制线粒体钙内流,进而抑制 FDXR-FDX1-LIAS 通路介导的 DLST 硫辛酰化与铜死亡,从而保护血管平滑肌细胞并减缓 AAD 进展。该研究不仅深化了对主动脉疾病发生机制的认识,也为开发靶向铜死亡与代谢稳态的新型联合干预策略提供了重要的理论基础。
复旦大学附属中山医院心内科任骏教授与张英梅教授为本文的共同通讯作者。复旦大学附属中山医院心内科林洁博士、熊晟均博士和安颖博士为本文的共同第一作者。该研究工作得到葛均波院士的大力支持,并受到国家重点研发计划、国家自然科学基金杰青及重点等项目的资助。
论文链接:
https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.125.327965
文章来源:论道心血管









