

关键词:
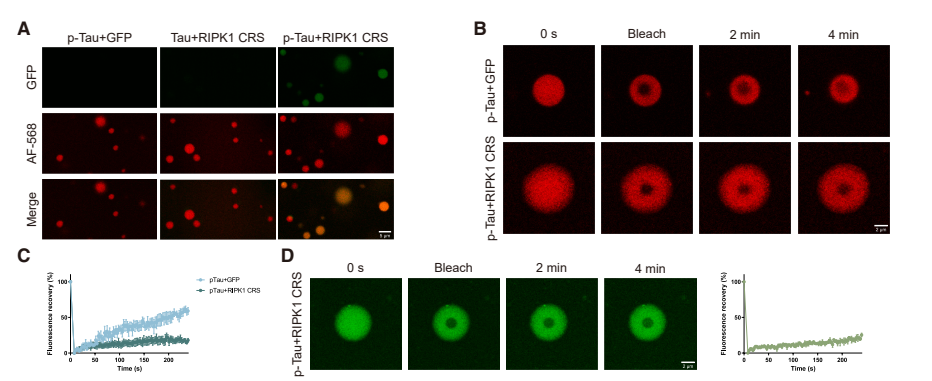
阿尔茨海默病患者的大脑中,过度磷酸化的 Tau 蛋白往往比临床症状早出现数十年,但为何神经元要在 Tau 积累多年后才死亡?这个长期困扰神经科学界的谜题暗示,Tau 病理本身可能只是「伏笔」,真正的神经元死亡需要另一个关键因素配合。 2026 年 4 月 22 日,中国科学院上海有机化学研究所袁均瑛院士、邹成宇及刘聪团队联合德国慕尼黑大学 Jochen Herms 团队在顶级期刊 Neuron 杂志在线发表题为「Glucose hypometabolism and hyperphosphorylated Tau synergistically drive neuronal necroptosis」的研究论文。这项研究不仅发现葡萄糖低代谢与过度磷酸化 Tau 蛋白协同通过坏死性凋亡(necroptosis)途径杀死神经元,更揭示了 A20 分子检查点失效和受体相互作用蛋白激酶 1(receptor-interacting protein kinase 1,RIPK1)蛋白结合的关键机制,还发现常见的膳食补充剂乙酰左旋肉碱(acetyl-L-carnitine,ALCAR)具有显著的治疗潜力。 双重打击模型:Tau 提供「死亡平台」,葡萄糖缺乏解除「安全锁」 传统观点认为,Tau 蛋白缠结一旦形成就会直接导致神经元死亡,但临床观察显示许多老年人脑内有大量 Tau 病理却认知正常。研究团队推测,Tau 蛋白可能需要一个「帮凶」才能完成致命一击。他们利用表达突变 Tau 的细胞和小鼠模型,系统测试了不同葡萄糖浓度条件下的神经元存活情况。结果发现,在正常葡萄糖水平下,即使存在大量 p-Tau,神经元仍能存活;但在低葡萄糖条件下,p-Tau 阳性神经元迅速死亡,且这种死亡中凋亡或铁死亡抑制剂仅提供有限保护,而坏死性凋亡抑制剂挽救效果显著。这提示葡萄糖低代谢和 p-Tau 构成了杀死神经元的「双重打击」——p-Tau 提供「死亡平台」,葡萄糖缺乏解除「安全锁」。研究进一步证明,这一坏死性凋亡过程并不依赖经典的 TNF/TNFR1 信号通路,而是由低糖环境下的 p-Tau-RIPK1 异常结合直接驱动,这也是该研究的重要创新点之一。 刹车失效:A20 乙酰化降低,坏死性凋亡通路因此失控 有意思的是,研究团队进一步追踪了这一过程的分子开关。他们发现低葡萄糖条件下,一种名为 A20(TNFAIP3,坏死性凋亡负调控因子)的蛋白表达显著下降。A20 本是坏死性凋亡的「刹车」,但葡萄糖缺乏导致乙酰辅酶 A 减少,A20 的乙酰化修饰降低,使其被溶酶体降解。当 A20 水平下降,坏死性凋亡通路就失控了。更令人振奋的是,研究人员给小鼠喂食乙酰左旋肉碱 —— 一种常见的膳食补充剂,可以补充乙酰辅酶 A,恢复 A20 的乙酰化和稳定性。接受 ALCAR 治疗的阿尔茨海默病模型小鼠不仅显著缓解海马萎缩、神经元死亡减少,在空间记忆测试中也明显改善。 研究还发现,低葡萄糖环境不仅是外部代谢压力,还会进一步加重 Tau 病理本身:在低糖条件下,p38 MAPK 信号被激活,促使 Tau 磷酸化水平进一步升高,从而增强其致病能力。为了进一步解析 p-Tau 如何启动死亡信号,研究团队利用邻近标记技术鉴定了 p-Tau 的相互作用蛋白,发现坏死性凋亡的核心分子 RIPK1 与 p-Tau 直接结合。通过结构生物学分析,他们锁定 RIPK1 中间结构域的一段带电残基序列(CRS)是关键结合位点,p-Tau 的磷酸基团与这段序列形成静电相互作用。在 A20 下调的背景下,当 RIPK1 结合到 p-Tau 上,会被招募到 p-Tau 形成的液滴凝聚物中,在那里被激活并引发下游的坏死性凋亡级联反应。研究人员设计了阻断这一相互作用的竞争性多肽,发现它可以有效保护神经元免于死亡。 从机制到药物:ALCAR 补充与 RIPK1 阻断,两条干预路径 在此基础上,研究团队通过通过 AAV 介导的体内表达实验验证了靶向 RIPK1 的治疗潜力。他们利用腺相关病毒向阿尔茨海默病小鼠海马递送缺失 CRS 结构域的 RIPK1 突变体,这种突变体无法与 p-Tau 结合。结果显示,接受基因治疗的小鼠海马结构得到保护,认知功能显著改善,坏死性凋亡标志物水平大幅下降。这一结果不仅证明了 p-Tau-RIPK1 相互作用在体内的功能重要性,也为开发基于阻断这一相互作用的小分子药物或基因疗法提供了概念验证。 这项研究极大地改变了我们对阿尔茨海默病神经元死亡机制的理解。它打破了「Tau 缠结直接致死」的简单认知,揭示了一个需要代谢应激和蛋白病理协同作用的复杂调控网络,为理解为何葡萄糖低代谢是认知衰退的最佳预测指标提供了分子解释。更重要的是,研究为临床干预提供了双重靶点 —— 既可以通过 ALCAR 等代谢干预恢复 A20 检查点,也可以开发阻断 p-Tau-RIPK1 结合的新型药物。论文还提出了一个值得关注的临床启发:未来阿尔茨海默病相关干预或许可以结合 p-Tau 检测与 FDG-PET 代谢影像,对「 既存在 Tau 病理、又伴随脑葡萄糖低代谢」的患者进行更精准分层,从而提高坏死性凋亡靶向治疗的获益概率。 不过,这项研究仍存在局限。体外实验使用的 Tau 磷酸化模式无法完全模拟体内阿尔茨海默病的复杂病理;ALCAR 和 RIPK1 干预虽显著但未能完全阻止神经退行性变,提示可能存在其他并行机制;这些发现从概念验证到临床应用,还需要在人体中验证最佳剂量、治疗窗口和长期安全性。 原文链接: 2026-04-23 18:00点击次数:57 关键词: 阿尔茨海默病患者的大脑中,过度磷酸化的 Tau 蛋白往往比临床症状早出现数十年,但为何神经元要在 Tau 积累多年后才死亡?这个长期困扰神经科学界的谜题暗示,Tau 病理本身可能只是「伏笔」,真正的神经元死亡需要另一个关键因素配合。 2026 年 4 月 22 日,中国科学院上海有机化学研究所袁均瑛院士、邹成宇及刘聪团队联合德国慕尼黑大学 Jochen Herms 团队在顶级期刊 Neuron 杂志在线发表题为「Glucose hypometabolism and hyperphosphorylated Tau synergistically drive neuronal necroptosis」的研究论文。这项研究不仅发现葡萄糖低代谢与过度磷酸化 Tau 蛋白协同通过坏死性凋亡(necroptosis)途径杀死神经元,更揭示了 A20 分子检查点失效和受体相互作用蛋白激酶 1(receptor-interacting protein kinase 1,RIPK1)蛋白结合的关键机制,还发现常见的膳食补充剂乙酰左旋肉碱(acetyl-L-carnitine,ALCAR)具有显著的治疗潜力。 双重打击模型:Tau 提供「死亡平台」,葡萄糖缺乏解除「安全锁」 传统观点认为,Tau 蛋白缠结一旦形成就会直接导致神经元死亡,但临床观察显示许多老年人脑内有大量 Tau 病理却认知正常。研究团队推测,Tau 蛋白可能需要一个「帮凶」才能完成致命一击。他们利用表达突变 Tau 的细胞和小鼠模型,系统测试了不同葡萄糖浓度条件下的神经元存活情况。结果发现,在正常葡萄糖水平下,即使存在大量 p-Tau,神经元仍能存活;但在低葡萄糖条件下,p-Tau 阳性神经元迅速死亡,且这种死亡中凋亡或铁死亡抑制剂仅提供有限保护,而坏死性凋亡抑制剂挽救效果显著。这提示葡萄糖低代谢和 p-Tau 构成了杀死神经元的「双重打击」——p-Tau 提供「死亡平台」,葡萄糖缺乏解除「安全锁」。研究进一步证明,这一坏死性凋亡过程并不依赖经典的 TNF/TNFR1 信号通路,而是由低糖环境下的 p-Tau-RIPK1 异常结合直接驱动,这也是该研究的重要创新点之一。 刹车失效:A20 乙酰化降低,坏死性凋亡通路因此失控 有意思的是,研究团队进一步追踪了这一过程的分子开关。他们发现低葡萄糖条件下,一种名为 A20(TNFAIP3,坏死性凋亡负调控因子)的蛋白表达显著下降。A20 本是坏死性凋亡的「刹车」,但葡萄糖缺乏导致乙酰辅酶 A 减少,A20 的乙酰化修饰降低,使其被溶酶体降解。当 A20 水平下降,坏死性凋亡通路就失控了。更令人振奋的是,研究人员给小鼠喂食乙酰左旋肉碱 —— 一种常见的膳食补充剂,可以补充乙酰辅酶 A,恢复 A20 的乙酰化和稳定性。接受 ALCAR 治疗的阿尔茨海默病模型小鼠不仅显著缓解海马萎缩、神经元死亡减少,在空间记忆测试中也明显改善。 研究还发现,低葡萄糖环境不仅是外部代谢压力,还会进一步加重 Tau 病理本身:在低糖条件下,p38 MAPK 信号被激活,促使 Tau 磷酸化水平进一步升高,从而增强其致病能力。为了进一步解析 p-Tau 如何启动死亡信号,研究团队利用邻近标记技术鉴定了 p-Tau 的相互作用蛋白,发现坏死性凋亡的核心分子 RIPK1 与 p-Tau 直接结合。通过结构生物学分析,他们锁定 RIPK1 中间结构域的一段带电残基序列(CRS)是关键结合位点,p-Tau 的磷酸基团与这段序列形成静电相互作用。在 A20 下调的背景下,当 RIPK1 结合到 p-Tau 上,会被招募到 p-Tau 形成的液滴凝聚物中,在那里被激活并引发下游的坏死性凋亡级联反应。研究人员设计了阻断这一相互作用的竞争性多肽,发现它可以有效保护神经元免于死亡。 从机制到药物:ALCAR 补充与 RIPK1 阻断,两条干预路径 在此基础上,研究团队通过通过 AAV 介导的体内表达实验验证了靶向 RIPK1 的治疗潜力。他们利用腺相关病毒向阿尔茨海默病小鼠海马递送缺失 CRS 结构域的 RIPK1 突变体,这种突变体无法与 p-Tau 结合。结果显示,接受基因治疗的小鼠海马结构得到保护,认知功能显著改善,坏死性凋亡标志物水平大幅下降。这一结果不仅证明了 p-Tau-RIPK1 相互作用在体内的功能重要性,也为开发基于阻断这一相互作用的小分子药物或基因疗法提供了概念验证。 这项研究极大地改变了我们对阿尔茨海默病神经元死亡机制的理解。它打破了「Tau 缠结直接致死」的简单认知,揭示了一个需要代谢应激和蛋白病理协同作用的复杂调控网络,为理解为何葡萄糖低代谢是认知衰退的最佳预测指标提供了分子解释。更重要的是,研究为临床干预提供了双重靶点 —— 既可以通过 ALCAR 等代谢干预恢复 A20 检查点,也可以开发阻断 p-Tau-RIPK1 结合的新型药物。论文还提出了一个值得关注的临床启发:未来阿尔茨海默病相关干预或许可以结合 p-Tau 检测与 FDG-PET 代谢影像,对「 既存在 Tau 病理、又伴随脑葡萄糖低代谢」的患者进行更精准分层,从而提高坏死性凋亡靶向治疗的获益概率。 不过,这项研究仍存在局限。体外实验使用的 Tau 磷酸化模式无法完全模拟体内阿尔茨海默病的复杂病理;ALCAR 和 RIPK1 干预虽显著但未能完全阻止神经退行性变,提示可能存在其他并行机制;这些发现从概念验证到临床应用,还需要在人体中验证最佳剂量、治疗窗口和长期安全性。 原文链接: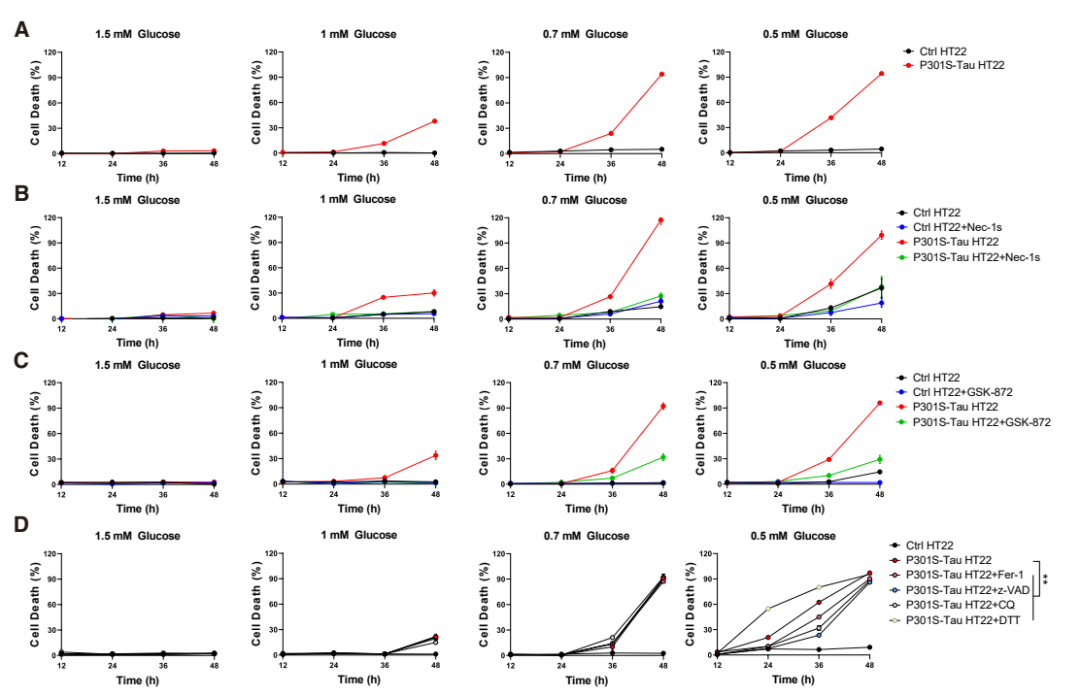
Neuron:阿尔茨海默病神经元死亡机制新解,袁均瑛院士等团队发现葡萄糖低代谢与 Tau 蛋白共同触发坏死性凋亡
https://doi.org/10.1016/j.neuron.2026.03.035









