

关键词:外泌体技术服务、外泌体提取鉴定
胶质母细胞瘤(GBM)是恶性程度最高的中枢神经系统肿瘤,预后极差、极易复发,核心驱动因素为脑肿瘤干细胞(BTSCs)。缺氧是胶质瘤微环境核心特征,可通过外泌体介导细胞间通讯,促进侵袭、转移与治疗抵抗。传统miRNA主要在胞质抑制基因表达,而核激活miRNA(NamiRNA)可入核结合增强子激活转录,但其在BTSCs中的作用尚不清楚。临床发现miR‑151a‑3p与多种肿瘤进展相关,但其是否通过外泌体传递、并以NamiRNA方式调控胶质瘤恶性表型仍未知。本研究围绕缺氧BTSCs外泌体miR‑151a‑3p展开,旨在揭示其核调控机制与促癌通路,为GBM提供全新预后标志物与治疗靶点。

今天分享的是一篇发表在【Cancer Letters】(IF:10.1)上题为“BTSCs exosomes derived NamiRNA-enhancer network of miR-151a-3p mediates a positive feedback loop and promotes the progression of glioma via FAK phosphorylation”的研究。本研究发现,缺氧BTSCs会通过外泌体释放一种名为miR-151a-3p的小分子RNA,它能进入癌细胞核发挥“核激活miRNA(NamiRNA)”作用,激活PDE4D基因,并形成正反馈循环,持续放大促癌信号。机制上,miR-151a-3p经由PDE4D/FAK/YAP通路,大幅增强胶质瘤的侵袭、转移和上皮间质转化,让肿瘤更恶性、更易复发。更重要的是,研究首次发现已上市药物Capmatinib能直接结合并抑制PDE4D,切断这条恶性通路,在动物模型中有效抑制肿瘤生长、延长生存期。这项成果揭示了胶质瘤恶化的全新机制,并提出miR-151a-3p/PDE4D可作为诊断标志物和治疗靶点,为脑胶质瘤提供了突破性的精准治疗新策略。
研究思路
1.表型初探:明确缺氧BTSCs外泌体(H‑exo)可促进胶质瘤增殖、侵袭、EMT与体内成瘤,说明外泌体是关键通讯介质。
2.分子筛选:筛选并验证外泌体中关键效应分子miR‑151a‑3p,证实其高表达于缺氧外泌体并可传递至受体细胞。
3.核机制解析:阐明miR‑151a‑3p作为NamiRNA,通过P300/H3K27ac激活PDE4D增强子,揭示全新核调控模式。
4.通路验证:证实PDE4D/FAK/YAP通路介导下游恶性表型,并形成miR‑151a‑3p‑PDE4D正反馈环。
5.转化探索:筛选并验证Capmatinib可直接结合PDE4D,阻断该轴以抑制胶质瘤进展。
研究创新性
1.科学问题创新
首次将缺氧BTSCs外泌体与核激活miRNA(NamiRNA)机制相结合,填补了胶质瘤干细胞外泌体在细胞核水平调控基因表达的研究空白。
2.分子机制创新
首次揭示外泌体miR‑151a‑3p作为NamiRNA,通过P300/H3K27ac乙酰化修饰激活PDE4D增强子,并形成miR‑151a‑3p‑PDE4D正反馈环路,颠覆传统miRNA仅在胞质抑制基因的认知。
3.信号轴创新
首次构建并证实NamiR‑151a‑3p/PDE4D/FAK/YAP全新促癌信号轴,阐明缺氧微环境驱动胶质瘤侵袭、转移与EMT的完整分子链条。
4.临床转化创新
首次发现已上市药物Capmatinib可直接靶向结合PDE4D,阻断上述促癌轴并抑制胶质瘤进展,为GBM提供“老药新用”的快速转化策略。
5.标志物创新
首次提出miR‑151a‑3p/PDE4D可作为胶质瘤预后判断与分级的新型分子标志物,为临床精准分型提供依据。。
研究成果
1.缺氧BTSCs来源的外泌体促进体内外胶质瘤进展
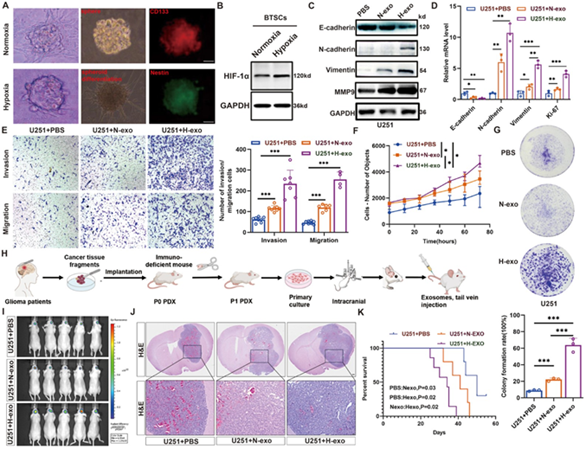
图1A:常氧与缺氧条件下BTSCs球形成及干细胞标志物CD133、Nestin表达。图1B:Western blot检测缺氧条件下BTSCs中HIF‑1α显著升高。
图1C:H‑exo处理后U251细胞EMT标志N‑cadherin、Vimentin、MMP9升高,E‑cadherin降低。
图1D:qRT-PCR验证EMT及增殖标志mRNA水平变化。
图1E:Transwell显示H‑exo显著增强侵袭与迁移。
图1F:HCS动态计数显示H‑exo促进细胞增殖。
图1G:克隆形成实验证实H‑exo提升克隆能力。
图1H:PDX原位胶质瘤模型构建流程。
图1I:活体成像显示H‑exo组肿瘤荧光更强。
图1J:HE显示H‑exo组肿瘤侵袭性更高、核分裂象增多。
图1K:H‑exo组小鼠生存期显著缩短。
这些结果表明,缺氧BTSCs来源的外泌体可显著促进胶质瘤细胞EMT、侵袭、增殖与体内成瘤,并缩短宿主生存期。
2. BTSCs来源的外泌体miR-151a-3p促进胶质瘤进展
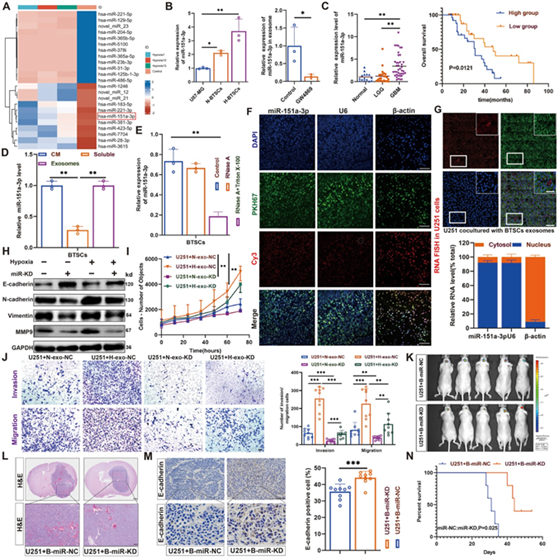
图2A:miRNA测序热图显示miR‑151a‑3p在H‑exo中显著上调。
图2B:qRT-PCR证实miR‑151a‑3p在缺氧BTSCs中高表达。
图2C:临床组织显示miR‑151a‑3p高表达与患者更差生存相关。
图2D:miR‑151a‑3p主要位于外泌体组分。
图2E:RNase+TritonX‑100处理可降解外泌体miR‑151a‑3p。
图2F:FISH显示miR‑151a‑3p主要定位于细胞核。
图2G:荧光示踪显示BTSCs来源的外泌体可被U251摄取。
图2H:敲低miR‑151a‑3p可逆转EMT标志物水平的改变。
图2I:HCS显示miR‑151a‑3p敲低抑制增殖。
图2J:Transwell显示敲低miR‑151a‑3p抑制侵袭迁移。
图2K:原位成像显示miR‑KD组肿瘤荧光减弱。
图2L:HE显示miR‑KD组肿瘤边界更清晰。
图2M:IHC显示miR‑KD组E‑cadherin回升,伴随着肿瘤侵袭和增殖减少。
图2N:miR‑KD组小鼠生存期显著延长。
这些结果表明,H-BTSCs来源的外泌体miR‑151a‑3p是介导缺氧BTSCs促癌作用的关键功能分子,可被传递至胶质瘤细胞核并促进恶性进展。
3. P300介导H3K27ac乙酰化激活PDE4D增强子
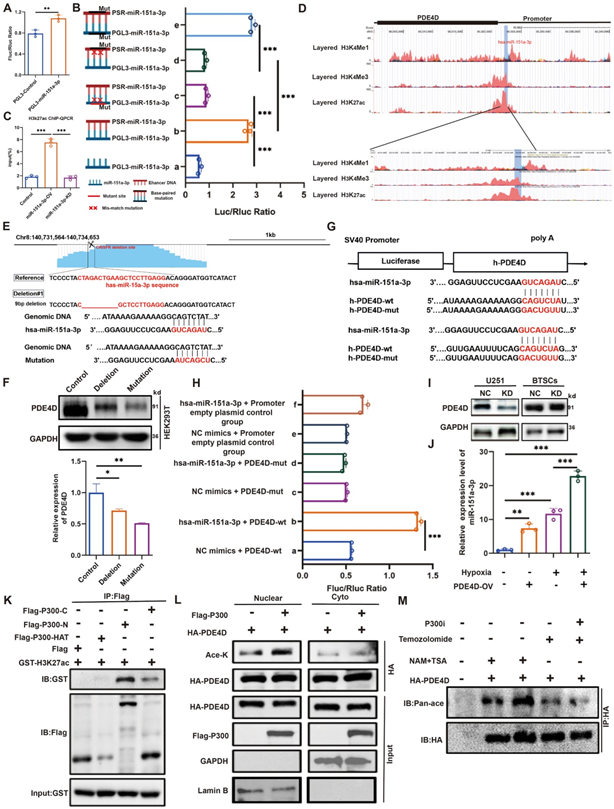
图3A:双荧光素酶证实miR‑151a‑3p靶位点具有增强子活性。
图3B:突变miR‑151a‑3p或增强子结合位点可消除荧光素酶激活。
图3C:ChIP‑qPCR显示miR‑151a‑3p促进H3K27ac富集。
图3D:UCSC显示该区域富集激活型增强子信号。
图3E/F:CRISPR敲除增强子后PDE4D无法被miR‑151a‑3p激活,而敲除增强子显著降低PDE4D表达。
图3G:PDE4D启动子突变位点示意图。
图3H:荧光素酶证实miR‑151a‑3p结合PDE4D启动子。
图3I:敲低miR‑151a‑3p降低PDE4D蛋白表达。
图3J:过表达PDE4D可反过来上调miR‑151a‑3p。
图3K/L:P300截断体结合实验显示H3K27ac特异性地与P300的N端和C端区域结合,并且P300主要在细胞核促进PDE4D乙酰化。
图3M:去乙酰化酶抑制剂进一步提升PDE4D乙酰化。
这些结果表明,外泌体miR‑151a‑3p作为NamiRNA入核,通过P300/H3K27ac通路激活PDE4D增强子,并与PDE4D形成正反馈环。
4.外泌体miR‑151a‑3p靶向PDE4D促进胶质瘤进展
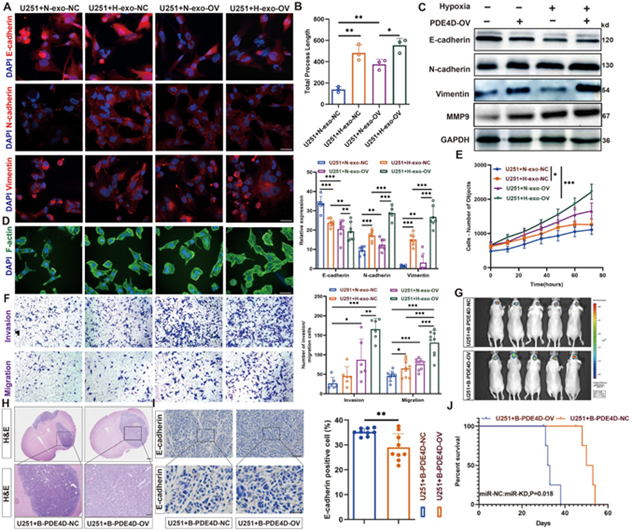
图4A/C:IF显示PDE4D过表达促进EMT标志物水平的变化。
图4B/D:FITC‑Phalloidin显示缺氧协同PDE4D促进细胞骨架重构与突起延长。
图4E:HCS显示PDE4D显著促进增殖。
图4F:Transwell显示PDE4D增强侵袭迁移。
图4G:原位成像显示PDE4D过表达加速肿瘤生长。
图4H:HE显示PDE4D过表达组肿瘤侵袭性增强。
图4I:IHC显示PDE4D过表达组E‑cadherin降低。
图4J:PDE4D过表达显著缩短生存期。
这些结果表明,PDE4D是miR‑151a‑3p的关键下游靶基因,可强烈促进胶质瘤恶性表型与体内进展,尤其是在缺氧条件下。
5.外泌体miR‑151a‑3p通过PDE4D/FAK/YAP通路诱导EMT
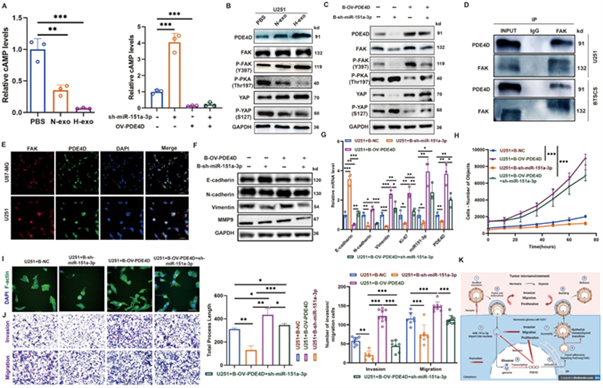
图5A:H‑exo与PDE4D过表达均降低U251细胞内cAMP水平。
图5B/C:Western blot显示PDE4D激活FAK磷酸化、抑制PKA、促进YAP去磷酸化。
图5D:Co‑IP证实PDE4D与FAK直接结合。
图5E:IF显示PDE4D与FAK在胞质中共定位。
图5F/G:rescue实验显示敲低miR‑151a‑3p可逆转PDE4D诱导的EMT。
图5H:HCS实验显示miR‑151a‑3p敲低可逆转PDE4D促增殖作用。
图5I/J:miR‑151a‑3p敲低可逆转PDE4D促侵袭迁移作用。
这些结果表明,源自H-exo的miR‑151a‑3p通过PDE4D/FAK/YAP轴,以cAMP/PKA依赖方式促进YAP激活,最终驱动EMT与侵袭(图5K)。
6. Capmatinib直接结合PDE4D在体内外抑制胶质瘤进展
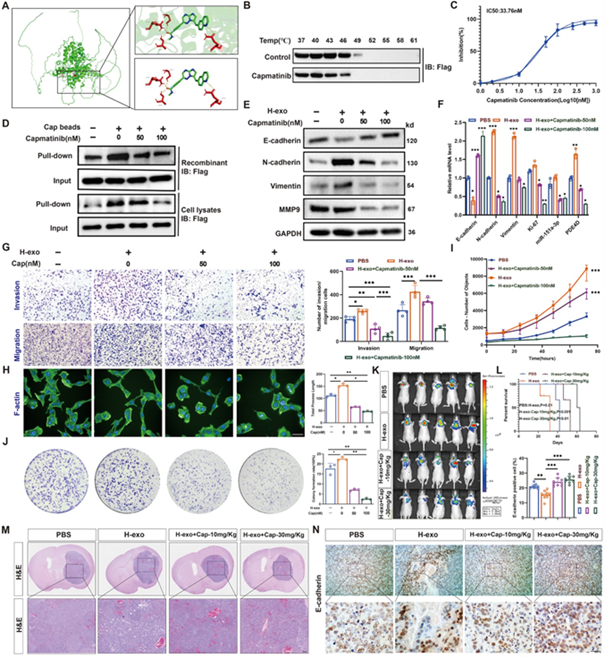
图6A:分子对接显示Capmatinib结合PDE4D的多个活性口袋。
图6B:CETSA显示Capmatinib可结合并稳定PDE4D。
图6C:TR‑FRET显示Capmatinib抑制PDE4D酶活(IC50=33.76nM)。
图6D:Pull‑down证实Capmatinib特异性结合PDE4D,并被过量Capmatinib以浓度依赖性方式竞争性阻断。
图6E/F:Capmatinib逆转H‑exo诱导的EMT并降低miR‑151a‑3p/PDE4D。
图6G/H/I/J:Capmatinib逆转H-exo诱导的细胞侵袭迁移、极化和增殖。
图6K:原位成像显示Capmatinib抑制肿瘤生长。
图6L:Capmatinib显著延长生存期。
图6M/N:HE与IHC显示Capmatinib降低侵袭和增殖并恢复E‑cadherin表达。
这些结果表明,Capmatinib(体内具有良好生物安全和低毒性)可直接靶向结合PDE4D,阻断miR‑151a‑3p/PDE4D/FAK/YAP轴,有效抑制胶质瘤恶性进展。
研究结论
本研究首次发现缺氧脑肿瘤干细胞通过外泌体传递NamiR‑151a‑3p,该miRNA进入胶质瘤细胞核后作为核激活miRNA,经P300介导H3K27ac乙酰化激活PDE4D增强子,形成miR‑151a‑3p‑PDE4D正反馈环路,并通过PDE4D/FAK/YAP通路促进EMT、侵袭、增殖与体内成瘤。此外,进一步筛选发现FDA获批药物Capmatinib可直接结合并抑制PDE4D,有效阻断该促癌轴,抑制肿瘤生长,具有良好的胶质瘤治疗潜力。该研究揭示了胶质瘤缺氧微环境中全新的外泌体‑NamiRNA调控机制,为GBM提供了miR‑151a‑3p/PDE4D这一高价值预后标志物与精准治疗新靶点。









