

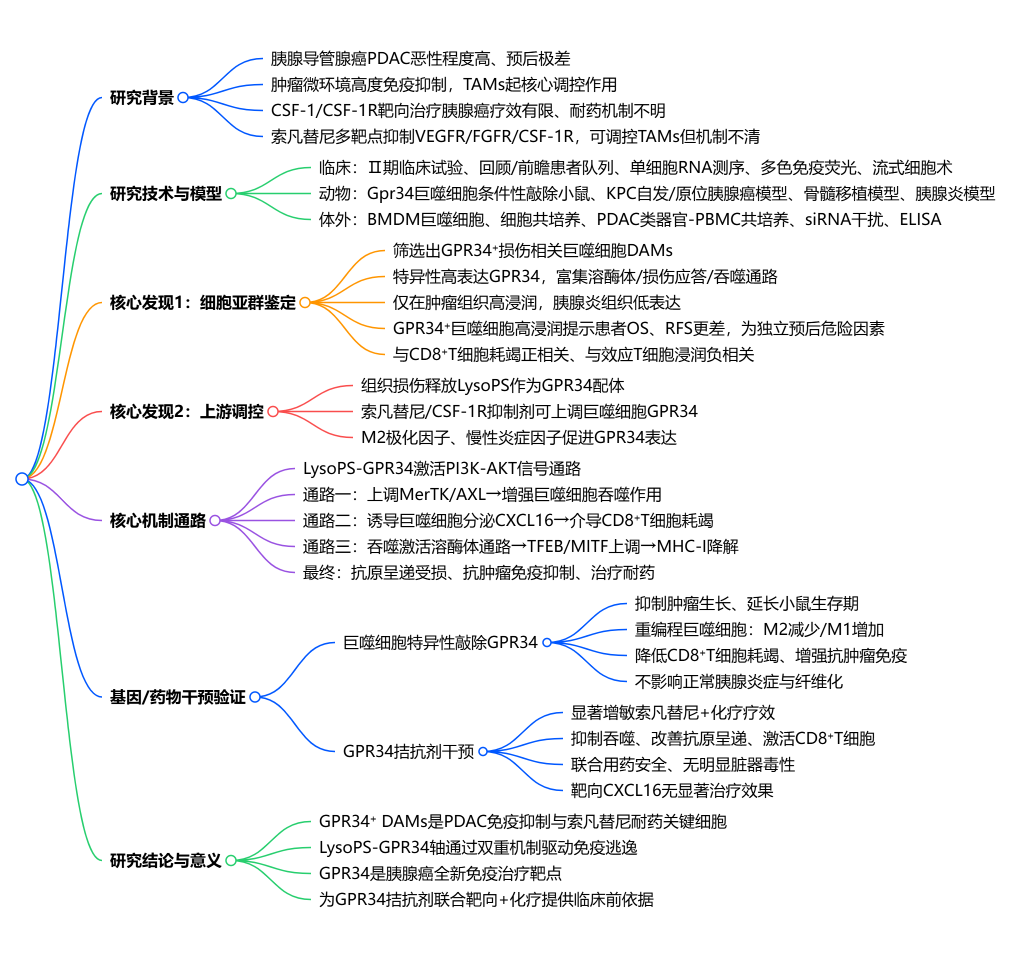
关键词:基因组表观组微生物组代谢组单细胞转录组空间转录组蛋白组
胰腺导管腺癌(PDAC)具有极强侵袭性与致死性,免疫抑制性TME是预后差、治疗耐药的核心原因;肿瘤相关巨噬细胞(TAMs)是介导 PDAC 免疫逃逸、化疗耐药的关键细胞。靶向 CSF-1/CSF-1R 通路可调控 TAMs,但在 PDAC 中疗效有限、耐药机制不明。索凡替尼(Surufatinib) 是多靶点激酶抑制剂,可抑制 VEGFR、FGFR、CSF-1R,能调控 TAMs 并重塑 TME,但其联合化疗在 PDAC 中的免疫调控机制及耐药诱因尚未阐明。 2026年4月28日,天津医科大学研究团队在国际知名期刊Signal Transduction and Targeted Therapy上发表了题为“Targeting GPR34 in damage-associated macrophages enhances anti-tumor immunity and the efficacy of Surufatinib in pancreatic cancer”的研究成果。 研究团队揭示GPR34⁺ DAMs是 PDAC 免疫抑制微环境和索凡替尼联合化疗耐药的关键驱动细胞。LysoPS-GPR34 轴通过CXCL16 分泌诱导 T 细胞耗竭、溶酶体通路降解 MHC-I 削弱抗原呈递双重机制,介导巨噬细胞免疫抑制与治疗耐药。靶向抑制 GPR34 可重编程 TAMs、恢复 CD8⁺T 细胞抗肿瘤免疫,显著提升索凡替尼联合化疗的抗胰腺癌疗效,GPR34 可作为 PDAC 免疫治疗全新靶点。该研究揭示了 PDAC 中巨噬细胞亚群异质性及索凡替尼治疗耐药新机制,阐明 LysoPS-GPR34 调控吞噬作用与肿瘤免疫的分子通路,为 PDAC 开发GPR34 靶向联合的综合治疗方案提供了坚实的临床前理论与实验依据。
·研究思路·
·研究结果·
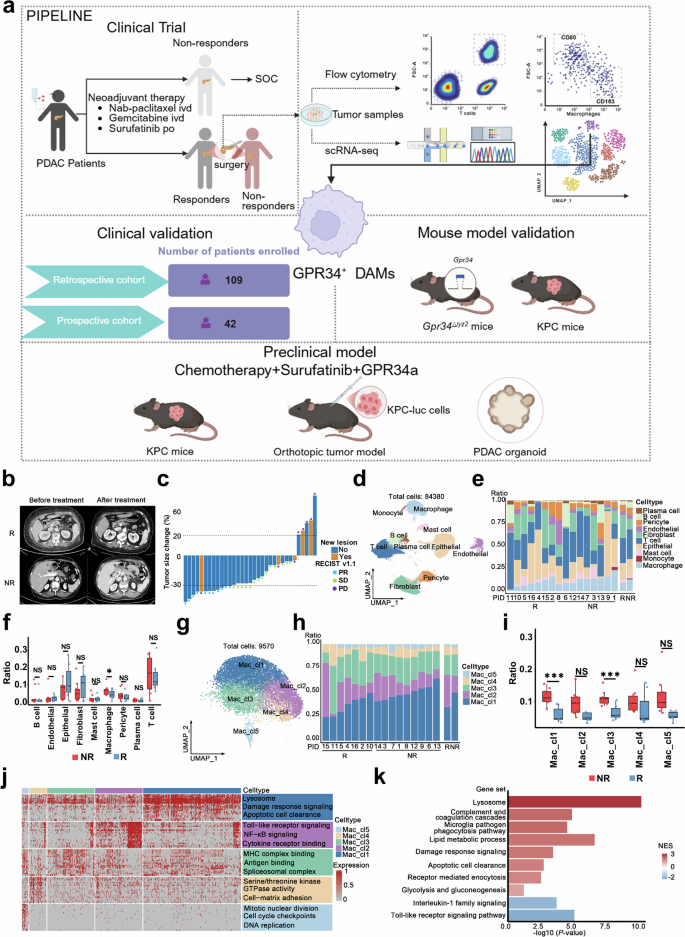
TCGA 数据库分析显示:CSF1-CSF1R 通路在胰腺癌组织中特异性上调,而 FGFR1、VEGFA 主要在正常胰腺组织表达;公共单细胞数据库证实 CSF1R 在 PDAC 单核 / 巨噬细胞中特异性高富集。鉴于 CSF-1R 在胰腺癌患者中显著高表达,推测索凡替尼可通过抑制巨噬细胞 CSF-1R 通路发挥免疫调控作用。为阐明 CSF-1R 抑制对肿瘤免疫微环境的影响及索凡替尼耐药机制,对入组患者肿瘤样本开展单细胞 RNA 测序,并在回顾性/前瞻性临床队列、体内外模型及临床前转化模型中进行验证。依据 RECIST 1.1 影像学标准将患者分为治疗有效组与无效耐药组:达到部分缓解(PR)或疾病稳定(SD)且肿瘤缩小归为有效组;疾病进展(PD)或疾病稳定但肿瘤增大归为无效组。 降维聚类分析显示:有效组与无效组的免疫细胞、上皮细胞组成存在显著差异;无效组巨噬细胞比例显著升高。鉴于索凡替尼靶向巨噬细胞 CSF1R,进一步分析两组巨噬细胞亚群差异。巨噬细胞重聚类为5 个亚群,其中Mac_cl1 亚群在治疗无效组显著富集。GSEA 富集分析显示 Mac_cl1 高富集溶酶体、损伤应答、凋亡细胞清除,提示该亚群参与免疫抑制微环境重塑。由于 Mac_cl1 在无效组占比极高(40%–60%),推测该亚群是介导索凡替尼联合化疗耐药的关键群体。这些结果表明:高富集溶酶体与吞噬通路的 Mac_cl1 巨噬细胞与索凡替尼治疗耐药高度相关。 图1 单细胞RNA测序揭示了化疗联合舒鲁替尼治疗后胰腺癌的细胞组成变化1、巨噬细胞重编程与胰腺癌索凡替尼治疗耐药相关
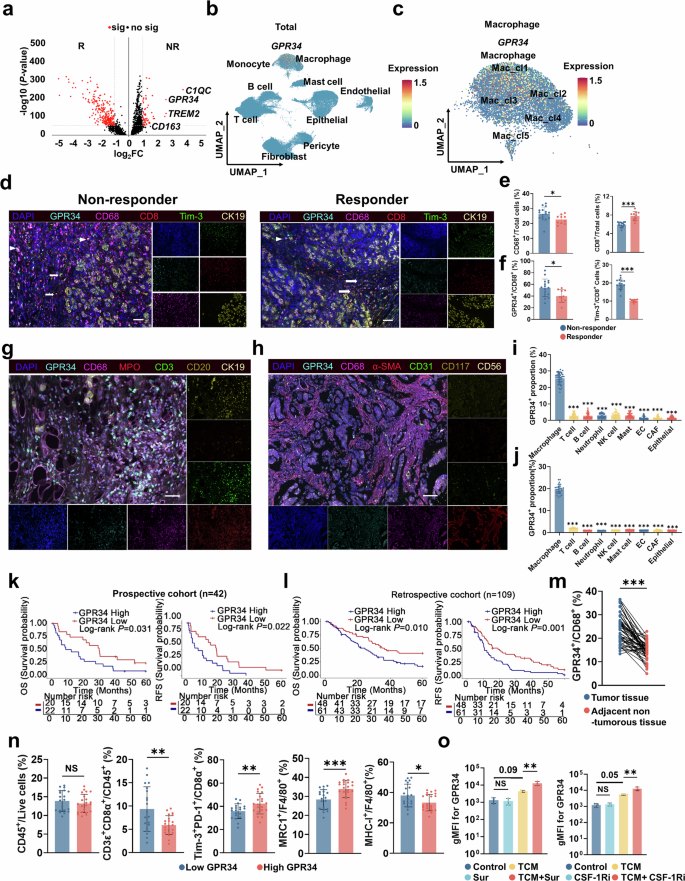
单细胞测序证实 Mac_cl1 是一类特征明确的损伤相关巨噬细胞 DAMs,高表达组织损伤应答与修复相关转录因子,高补体活化信号、高 C1q 家族基因表达,是修复型巨噬细胞的典型特征。分析 Mac_cl1 凋亡细胞清除与损伤识别受体表达发现:高表达 TREM2、CD163、ADORA3、SLAMF8、CD180 及GPR34,且呈现典型 M2 样极化特征。其中 GPR34、CD163、TREM2 在无效组显著上调;且GPR34 特异性局限于 Mac_cl1。GPR34 是溶血磷脂酰丝氨酸(LysoPS)的特异性受体,LysoPS 在坏死组织及胰腺癌微环境中显著升高。mIF 证实:无效组GPR34⁺CD68⁺巨噬细胞浸润显著高于有效组;而 TREM2⁺巨噬细胞两组无差异,提示GPR34是 DAMs 应答损伤信号的关键分子。同时无效组耗竭 CD8⁺T 细胞比例显著升高。 对回顾性队列 109 例、前瞻性队列 42 例PDAC 组织芯片分析显示:GPR34⁺巨噬细胞浸润比例与 CD8⁺T 细胞浸润负相关、与 Treg 浸润正相关;GPR34⁺巨噬细胞高浸润患者无复发生存(RFS)、总生存(OS)显著缩短;多因素 Cox 回归证实 GPR34⁺巨噬细胞高浸润是 OS、RFS 的独立危险因素。空间分布显示:GPR34⁺TAMs 主要局限于肿瘤巢内,CD8⁺T 细胞多分布于肿瘤间质周边,形成空间免疫隔离。这些结果表明GPR34⁺TAMs 与 PDAC 免疫抑制及不良预后密切相关。 为探究 GPR34 表达调控机制:采用肿瘤条件培养基 TCM 刺激小鼠骨髓来源巨噬细胞 BMDM,分别给予索凡替尼及特异性 CSF-1R 抑制剂;单独药物无法诱导 GPR34 上调,但药物联合肿瘤微环境因子可显著升高巨噬细胞 GPR34 表达;同时 M2 极化因子(IL-4、IL-13)及慢性炎症因子(IL-1β、IL-6)也可上调 GPR34。这些结果表明:索凡替尼介导的 CSF-1R 阻断会在肿瘤细胞因子微环境中进一步上调巨噬细胞 GPR34,GPR34 高表达与免疫抑制及治疗耐药密切相关。 图2 队列研究和体外研究揭示GPR34在巨噬细胞中的作用2、化疗诱导的损伤相关巨噬细胞(DAMs)驱动免疫抑制与治疗耐药
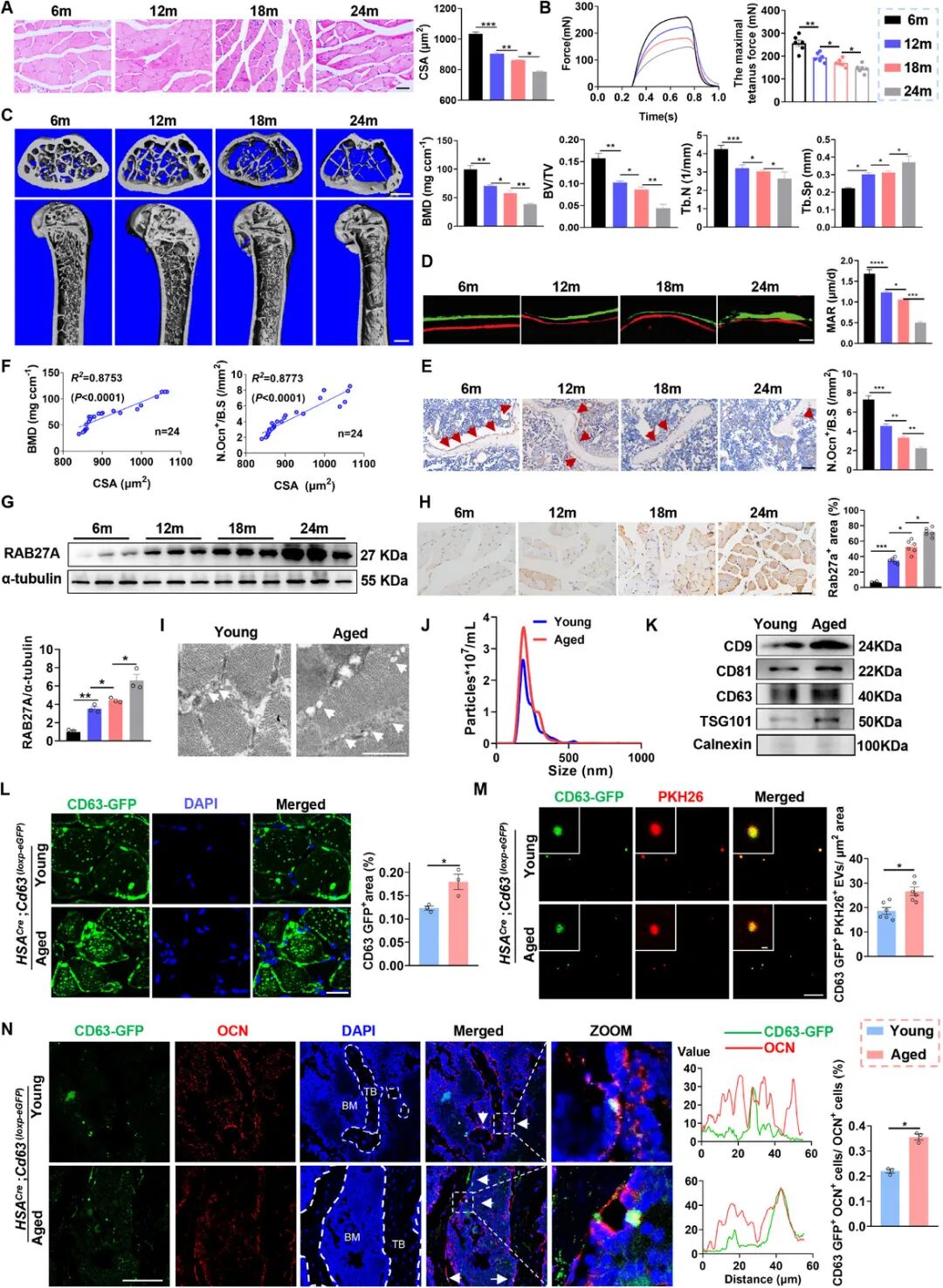
为明确 GPR34 在胰腺癌进展中巨噬细胞的功能,构建了Gpr34 敲除小鼠模型。活体成像显示:无化疗干预时,Gpr34 敲除与野生型小鼠肿瘤生长无差异;给予化疗后,Gpr34 敲除小鼠肿瘤负荷显著降低,提示 GPR34 可促进肿瘤对治疗损伤的免疫耐受。免疫谱分析显示:Gpr34 敲除后免疫抑制性巨噬细胞减少、效应 CD8⁺T 细胞增多、耗竭 T 细胞减少;巨噬细胞向 M1 极化偏移、M2 减少,树突细胞浸润增加,抗原呈递能力增强;MDSC、Treg、单核细胞等其他免疫细胞无明显变化,外周血免疫亚群也无显著改变。骨髓移植模型超声监测证实:GPR34 敲除可显著增强化疗抑瘤效果、延长小鼠生存期;组织染色显示肿瘤细胞凋亡增加、增殖降低;mIF 证实肿瘤微环境中 Tim-3⁺耗竭 CD8⁺T 细胞比例显著下降。 为评估 GPR34⁺巨噬细胞的吞噬能力,流式分选原位肿瘤 CD45⁺CD11b⁺F4/80⁺巨噬细胞,检测其吞噬 GFP 标记凋亡肿瘤细胞能力。结果显示:Gpr34 敲除小鼠巨噬细胞显著受损,吞噬凋亡肿瘤细胞比例明显降低。体外 BMDM 共培养实验同样证实:Gpr34 缺失巨噬细胞对化疗诱导凋亡肿瘤细胞的吞噬能力下降;流式检测相关受体 MerTK、AXL 表达显著下调。这些结果表明:GPR34 是胰腺癌中巨噬细胞介导吞噬作用与免疫抑制的关键调控分子;缺失 GPR34 可重塑肿瘤微环境、减弱巨噬细胞免疫抑制活性、促进 CD8⁺T 细胞浸润、抑制凋亡细胞清除。 图3 巨噬细胞特异性GPR34敲除可改善小鼠化疗效果3、GPR34 调控巨噬细胞介导的免疫抑制与吞噬作用
为阐明 GPR34⁺TAMs 介导免疫抑制的下游机制,在 Gpr34 敲除 / 野生小鼠体内分别清除 CD8⁺T 细胞与 NK 细胞。结果显示:清除 CD8⁺T 细胞可完全逆转 Gpr34 敲除带来的化疗增敏效应,而清除 NK 细胞无明显影响;提示 GPR34 主要依赖 CD8⁺T 细胞调控肿瘤杀伤。建立了 BMDM 与 CD8⁺T 细胞体外共培养体系:经化疗肿瘤条件培养基刺激后,Gpr34 敲除后 BMDM 免疫抑制活性下降、吞噬能力减弱、MHC-I 表达上调;伴随 CD8⁺T 细胞耗竭降低、细胞毒功能增强、效应 T 细胞分化增多。Gpr34 缺失 BMDM 可增强抗原特异性 CD8⁺T 细胞杀伤活性、降低耗竭水平;T 细胞杀伤实验进一步验证 GPR34 可通过巨噬细胞调控 CD8⁺T 细胞特异性杀瘤能力。这些结果证实:敲低 GPR34 可显著降低 CD8⁺T 细胞耗竭、增强细胞毒功能。 单细胞互作分析显示:GPR34⁺TAMs 与 T 细胞间CXCL、CCL 信号通路互作显著富集,其中CXCL16是 GPR34⁺巨噬细胞上调最显著的分泌型蛋白。Gpr34 敲除 BMDM 的 CXCL16 转录及蛋白分泌水平均显著降低;沉默 BMDM 中 Cxcl16 可逆转 LysoPS(GPR34 激动剂)诱导的 CD8⁺T 细胞耗竭、恢复其细胞毒功能。这些结果表明:GPR34 通过诱导巨噬细胞分泌 CXCL16,进而驱动 CD8⁺T 细胞耗竭,强化 PDAC 肿瘤微环境免疫抑制与化疗耐药。 图4 通过巨噬细胞和CD8+T细胞共培养证实了GPR34功能4、GPR34 通过巨噬细胞分泌 CXCL16 驱动 CD8⁺T 细胞耗竭
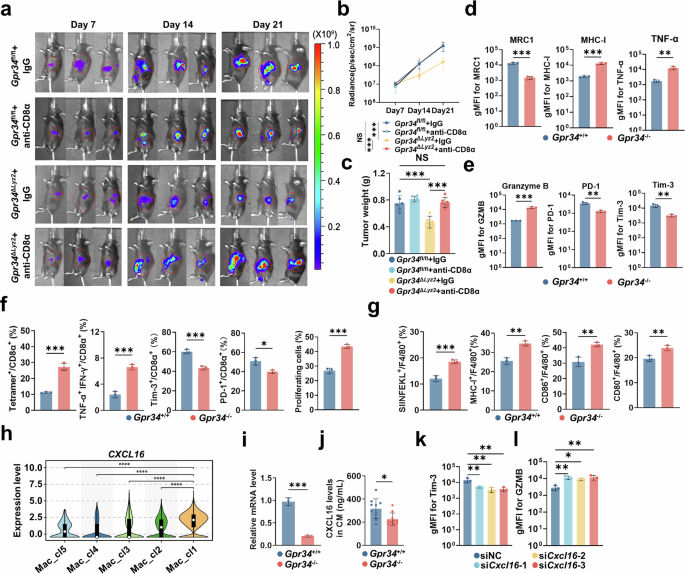
LysoPS 是 GPR34目前已知配体,LysoPS及其前体磷脂酰丝氨酸(PS)在组织损伤和细胞凋亡过程中均大量产生。LysoPS 处理可显著增强巨噬细胞吞噬作用,而 Gpr34 敲除后该效应显著减弱;LysoPS 通过 GPR34激活 PI3K-AKT 通路。LysoPS 经 GPR34 促进巨噬细胞免疫抑制表型、下调 MHC-I 表达;使用 PI3K/AKT 抑制剂可同时抑制巨噬细胞吞噬作用、逆转 LysoPS 诱导的免疫抑制。证实LysoPS-GPR34 轴通过 PI3K-AKT 通路介导巨噬细胞吞噬活化与免疫抑制。 单细胞分析显示 GPR34⁺TAMs 高表达 MERTK、AXL、TYRO3、GAS6 等吞噬相关基因;巨噬细胞 Gpr34 敲除可逆转这些凋亡识别受体的上调。体外实验证实LysoPS-GPR34 轴可通过 AKT 依赖方式上调 MerTK 表达,增强 PS 介导的吞噬作用;同时 LysoPS 可直接诱导免疫抑制因子 CXCL16 分泌。这些结果表明:组织损伤释放的 LysoPS 激活巨噬细胞 GPR34,经PI3K-AKT 通路上调 MerTK增强吞噬,同时直接促进 CXCL16 分泌,共同诱导 CD8⁺T 细胞耗竭。 图5 LysoPS-GPR34 调节巨噬细胞的吞噬作用和炎症细胞因子分泌5、LysoPS-GPR34 信号轴调控巨噬细胞吞噬与细胞因子分泌
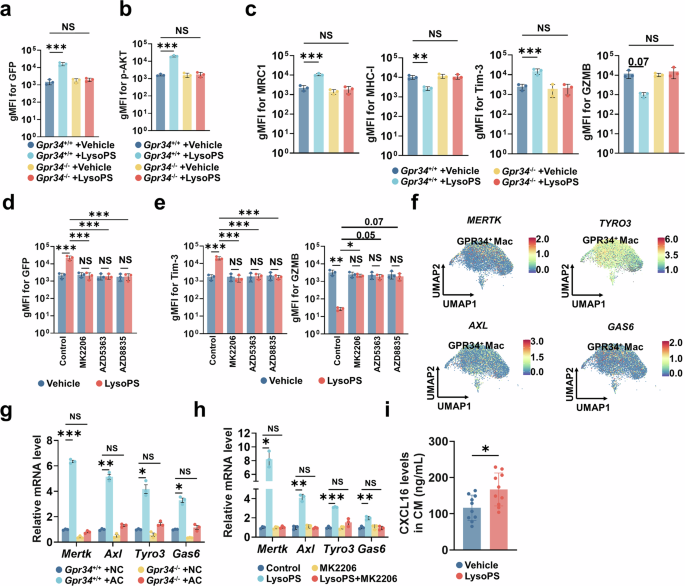
该研究发现巨噬细胞 GPR34 表达与 MHC-I 蛋白水平降低、抗原呈递能力受损相关;单独 LysoPS 处理不影响 MHC-I,但巨噬细胞与凋亡肿瘤细胞共培养后 MHC-I 显著下调,提示 MHC-I 抑制依赖吞噬作用。使用 MerTK 抑制剂可逆转吞噬介导的 MHC-I 下调,增强巨噬细胞抗原呈递、提升 T 细胞细胞毒功能、缓解 CD8⁺T 细胞耗竭。 单细胞测序显示 GPR34⁺TAMs 高表达溶酶体酶及相关基因;Gpr34 基因缺失可抑制溶酶体生成、阻断 MHC-I 降解。GPR34⁺TAMs 中溶酶体调控转录因子TFEB、MITF显著上调;沉默 TFEB/MITF 可阻断凋亡细胞摄取诱导的 MHC-I 降解、增强 CD8⁺T 细胞杀瘤功能。溶酶体抑制剂可破坏巨噬细胞持续性吞噬、阻断溶酶体介导的 MHC-I 降解,提升巨噬细胞向 CD8⁺T 细胞的抗原呈递能力。综上所述:LysoPS-GPR34 信号一方面诱导巨噬细胞分泌 CXCL16 导致 T 细胞耗竭;另一方面通过增强吞噬、激活溶酶体通路降解 MHC-I、损伤抗原呈递,双重通路驱动 CD8⁺T 细胞免疫耗竭。 图6 巨噬细胞吞噬功能通过MHC-I影响抗原呈递能力6、巨噬细胞吞噬通过溶酶体通路促进 MHC-I 降解
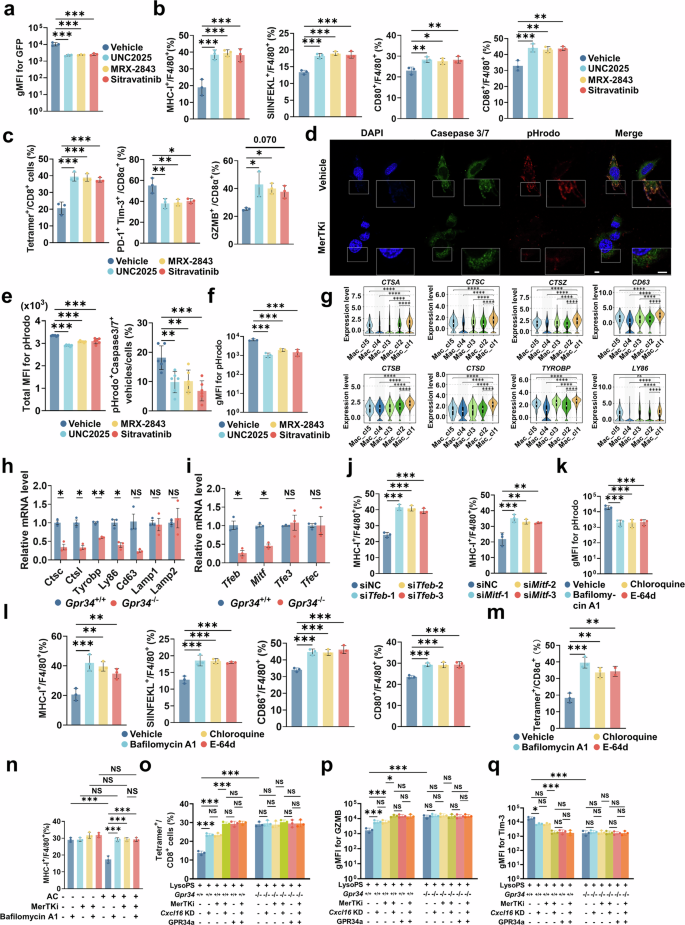
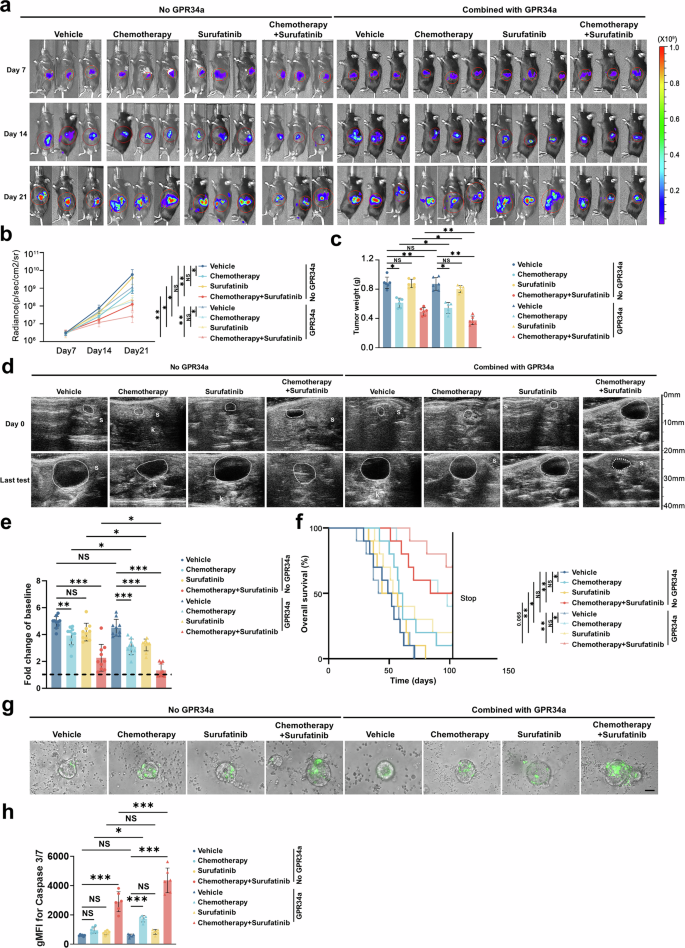
鉴于 LysoPS–GPR34 信号在吞噬、免疫抑制及索凡替尼耐药中的核心作用,在临床前模型中评估了 GPR34 拮抗剂的治疗潜力。胰腺原位模型显示:GPR34 拮抗剂可显著提升单纯化疗、单纯索凡替尼的抑瘤效果;三联方案(GPR34 拮抗剂 + 化疗 + 索凡替尼) 可强效抑制肿瘤生长、显著延长 KPC 小鼠生存期。患者肿瘤类器官-PBMC 共培养体系验证:GPR34 拮抗剂可稳定提升化疗及索凡替尼的抗肿瘤效果。安全性评价:三联用药小鼠心、肝、肺、肾等主要脏器无明显病理损伤;GPR34 拮抗剂联合索凡替尼不会加重化疗诱导的骨髓抑制及肝毒性,全身耐受性良好。进一步靶向下游 CXCL16:抗 CXCL16 治疗无法达到 GPR34 拮抗剂同等抑瘤效果,也无法显著增强 T 细胞细胞毒功能;提示相较于下游 CXCL16,上游 GPR34是更关键的免疫治疗靶点。这些结果表明:GPR34 拮抗剂可突破胰腺癌免疫抑制屏障、逆转治疗耐药、显著增强索凡替尼联合化疗疗效,是极具临床转化价值的联合治疗新策略。 图7 GPR34拮抗剂可增强化疗和索凡替尼的疗效7、GPR34 拮抗剂可增强索凡替尼与化疗治疗疗效
和元517·多组学嗨购季 

和元生物提供多组学服务(如转录组、蛋白组、代谢组、微生物组等)和单细胞及空间转录组服务(如10x Genomics GEM-X单细胞转录组(V4.0)、10x Genomics单细胞转录组(V3.1)、10x单细胞转录组及TCR/BCR、墨卓单细胞转录组、DNBelab C-TaiM4单细胞转录组、10x HD空间转录组等),致力于为广大生命科学家、医学工作者提供基于多组学的科研及临床应用解决方案。累计协助客户在Advanced Functional Materials、ACS Nano、Nature Aging、Cancer Research、Neuron、Advanced Science、Experimental & Molecular Medicine、Journal of Nanobiotechnology、Cell Death and Disease等期刊发表SCI论文,并推出了广受好评的生信分析云平台,助您高效实现个性化数据挖掘!和元生物CRO多组学服务









